No More Hindsight Bias: China’s Supreme People's Court Reins in Patent Inventiveness Assessments
For foreign applicants navigating patent litigation or invalidation in China, a common frustration is hindsight bias. Too often, CNIPA or lower courts strip down a patent into isolated technical features, find those features across disparate prior art references, and declare the invention to be "obvious". To counter such hindsight reasoning, the core criterion has always been whether a person skilled in the art would readily conceive of such technical solution.
In practice, that standard has not been applied consistently. Examiners and judges are often tempted to rely on their own intuitive standard, or worse-yet, directly use hindsight reasoning, undermining the consistency and predictability of the inventive-step analysis.
A relatively recent landmark decision by China’s Supreme People’s Court (SPC) (2024 Zhi Xing Zhong No. 7101 ”the 710 Case”) signals a welcome shift toward a more structured, objective, and predictable inventiveness assessment. In this case, the SPC provides a systematic explanation of how “technical teachings” should be identified. In particular, the SPC emphasized that the analysis should be conductedholistically by considering the relevant technical problem, application scenario, and technical effects. In doing so, the Court imposes important methodological constraints on inventive-step determinations.
The Facts: Same Flexible PCB but Different Uses
The central issue in the case was whether prior art reference “Evidence 14” —which disclosed the use of a flexible printed circuit board ("flexible PCB") in a pressure sensor—could provide a technical teaching for the distinguishing feature of the patent at issue, namely, using a flexible PCB in a vibration-and-shock composite sensor to isolate vibrations transmitted from the sensor housing to the interior of the sensor.
The conclusions diverged significantly depending on the stage of the proceedings:
Lower Instances: Both the CNIPA (invalidation stage) and the first-instance court ruled the invention to be obvious. Their reasoning was that flexible PCBs are well-known, so replacing one with another is an "obvious alternative".
SPC: The SPC reversed the decision, disagreeing that there existed a "technical teaching". The SPC emphasized that simply pointing out that a technical tool is "known" is not enough to prove that a person skilled in the art would naturally think of applying it to a completely different context.
Comparison of the Reasoning Adopted at Different Stages
To better understand the factors underlying these divergent conclusions, the key distinguishing technical features were analyzed as follows:
Stage | Core Conclusion | Reasoning | Methodology Issue |
Invalidation Proceeding | Obvious | Flexible PCB already known → could be substituted | Analogy based on technical means |
First Instance | Obvious | Motivation to improve + known technology | Motivation substituted for technical teaching |
Second Instance (SPC) | Not Obvious | Technical problem, application scenario, and technical effect all differ | Return to the technical context |
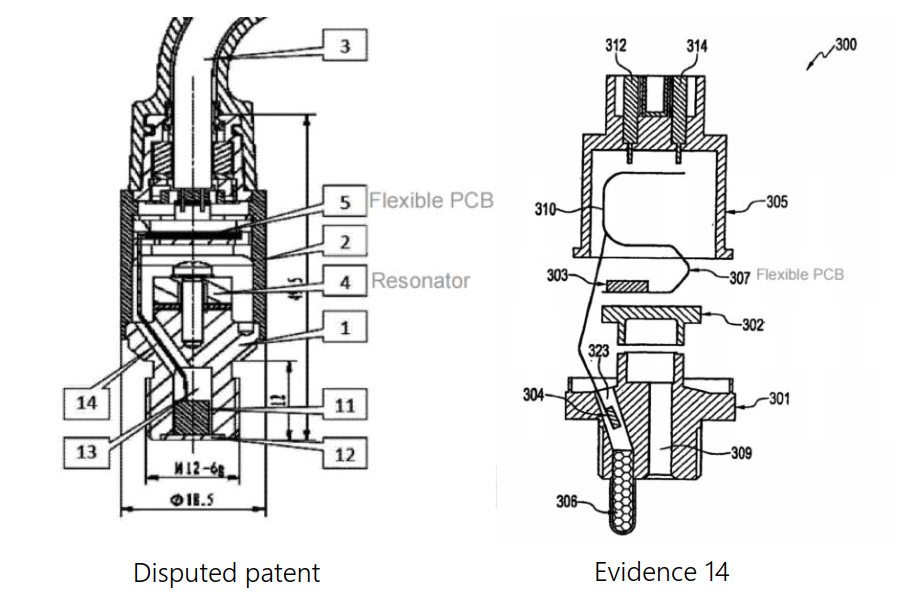
The SPC's conclusion was not based merely on the observation that both the patent and Evidence 14 employed a flexible PCB. Rather, the Court focused on the substantially different technical contexts in which the flexible PCB was used.
In Evidence 14, the flexible PCB serves as a connection medium for constructing a modular sensor architecture. The underlying technical problem is how to allow individual components to be positioned independently, rather than being fixed on a single rigid printed circuit board. By flexibly interconnecting components located at different positions, the flexible PCB enables the formation of a modular structure in which each module can be arranged at its optimal location.
By contrast, the patent addresses a fundamentally different problem arising in harsh operating environments such as rail transit and wind power generation. In these applications, vibration sensors are typically connected through large, rigid cables that must be secured by cable cleats. The substantial mass and rigidity of such cable assemblies transmit vibrations from the sensor housing directly to the internal resonator, leading to low-frequency resonance, frequency-response distortion, and degraded sensing performance. The technical problem addressed by the patent is therefore how to reduce resonance caused by the sensor's external structure and prevent vibration transmission from the housing to the internal sensing elements.
To solve this problem, the patent replaces conventional rigid cable connections with flexible PCB connections between sensor modules, so that vibration transmission from the outer housing to the interior can be reduced or isolated. As a result, low-frequency resonance interference is suppressed, and a flatter frequency response is restored.
Therefore, although both solutions employ a flexible PCB, the application scenarios, technical problems, and technical effects are materially different. Under such circumstances, the mere disclosure of a flexible PCB in Evidence 14 does not provide a technical teaching that would lead a person skilled in the art to adopt the claimed solution of the patent.
The Supreme Court’s Conclusion: Evidence 14 and the provided evidence of common knowledge did not disclose the distinguishing feature 1 of Flexible PCB, nor did they provide corresponding teaching. As a result, claim 1 as a whole was not disclosed by the prior art nor common knowledge, and thus possessed inventiveness.
Key Principles Established by the Supreme People's Court
- Prior Art Should be Considered “As a Whole”
Whether the prior art provides a technical teaching should be determined through a holistic assessment of the relevant technical problem, application scenario, and technical effects.
- Similar Technical Means Does not Equal a “Technical Teaching”
Just because a prior art reference has similar technical means does not mean that a “technical teaching” exists. For example, generally there is no technical teaching if the prior-art feature’s function differs from the function of the claimed invention’s distinguishing feature.
- Don’t Presume "Readily Conceivable" Without Supporting Evidence
If a feature has not been disclosed in the prior art and is not shown to be common general knowledge, one cannot presume that the feature is readily conceivable by a person skilled in the art.
- Technical Problem Should be Narrowed to a Specific Application
Define the technical problem based on the specific application environment and the role performed by the relevant structure, rather than generalizing at an abstract level.
Does the Prior Art Provide a Technical Teaching? A 3-Part Test
Based on the Court's reasoning, we can derive following three-factor analytical framework:
Same Technical Problem: Does the prior art address the same technical problem as the patent in suit?
Same Application Scenario: Are the respective technical fields and application environments the same or at least consistent?
Same Technical Effect: Does the technical feature achieve the same technical effect in both technical solutions?
Only when all three factors are satisfied should a technical teaching be established.
Eagle IP Commentary
The 710 Case together with related earlier cases reveal that the inventive-step assessment is changing from a conclusory form of reasoning to a structured analytical approach.
First, from the perspective of judicial methodology, this case clearly distinguishes three different approaches which focus on different aspects:
- analogy approach: the existence of similar technical means;
- motivation approach: the possibility or incentive for improvement;
- technical teaching approach: the specific technical context.
Through this decision, the SPC effectively imposed institutional limits on the first two approaches and made clear that inventive-step determinations must be based on verifiable technical teachings. This serves to curb the hindsight bias that has frequently appeared in practice.
Second, at the level of analytical reasoning, the concept of a “technical teaching” is further broken down into several constraining elements: the technical problem, the application scenario, and the technical effect. These factors form an integrated evaluative framework. Only when all factors are satisfied should a technical teaching be established.
This structural approach effectively changes the inquiry of whether a person skilled in the art "would have readily conceived" the claimed invention from a subjective cognitive assessment into an analysis that must consider objective technical facts. This standard should significantly enhance the transparency and predictability of inventive-step determinations in China.
Third, the case also raises the Examiner’s burden of proof for establishing that a claimed feature would have been “readily conceived”. For example, if the prior art does not disclose the relevant distinguishing feature, and there is insufficient evidence that the feature is “common general knowledge”, then the Examiner should not conclude a lack of inventive step solely based on “experience” or abstract reasoning.
Accordingly, this case provides a clearer direction for future inventive-step analysis. Rather than focusing on the abstract question of whether a skilled person "could have thought of" the claimed solution, the inquiry should return to a more concrete question: does the existing prior art contain a genuine technical teaching leading to the claimed invention?
If you would like to have more information on this matter or would like to have our advice, please feel free to contact us at [email protected].
This article is for general informational purposes only and should not be considered legal advice or a legal opinion on a specific set of facts.
- 最高人民法院行政判决书:(2024)最高法知行终710号。 ↩︎


